Chimica Farmaceutica - Chemical Biology. Coordinatrice: Roberti
Progettazione e sintesi di nuove piccole molecole bioattive da utilizzare sia nella ricerca di lead per lo sviluppo di nuovi agenti di interesse terapeutico che come probes per esplorare i diversi pathway biologici e le funzioni delle proteine target all’interno della cellula (Chemical Biology).
Temi di ricerca del gruppo
I principali temi di ricerca riguardano la progettazione e la sintesi di piccole molecole capaci di modulare l’attività di target molecolari coinvolti nello sviluppo tumorale. La progettazione razionale delle nuove molecole viene effettuata in collaborazione con il gruppo di chimica farmaceutica computazionale (Prof. F. Falchi e Prof. A Cavalli attualmente in aspettativa fino al 31/12/2026). Le strategie sintetiche applicate prevedono oltre a metodi “classici” un approccio di chimica combinatoria, operando sintesi in parallelo per ottenere piccole librerie di molecole drug-like che sono caratterizzate con tecniche spettroscopiche e spettrometriche. Inoltre vengono effettuate sintesi assistite da microonde nell’ambito di una chimica sostenibile. Di seguito sono descritti i due progetti principali.
Progettazione e sintesi di inibitori dell’interazione proteina-proteina Rad51/BRCA2
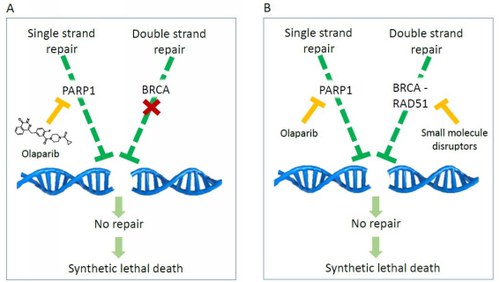
L’interazione Rad51/BRCA2 è fondamentale per la riparazione dei danni alla doppia elica del DNA (ricombinazione omologa (HR)). Dal momento che molti chemioterapici antitumorali causano danni al doppio filamento del DNA, inibire HR può aumentare la sensibilità delle cellule tumorali a questi chemioterapici e ad altri inibitori della riparazione dei danni al DNA, come ad esempio gli inibitori PARP. Olaparib è stato il primo inibitore PARP approvato nel 2014 per il trattamento di cancro al seno e tumori pancreatici con BRCA2 non funzionante. In questi pazienti, inibendo PARP vengono simultaneamente compromessi due meccanismi indipendenti di riparazione del DNA, inducendo così la letalità sintetica e la morte delle cellule tumorali (Fig. 1A). I nuovi inibitori Rad51-BRCA2 possono quindi essere studiati come agenti chemiosensibilizzanti in associazione con chemioterapici che danneggiano il DNA o alla radioterapia. In particolare potrebbero compromettere chimicamente il funzionamento di BRCA2 nei pazienti nei quali non è difettoso e in associazione con olaparib innescare una letalità sintetica (LS) totalmente generata da molecole di sintesi, aprendo nuove strade alla terapia antitumorale basata su PARP inibitori. Il nuovo paradigma potrebbe anche essere esteso ad altre coppie di geni coinvolti in diversi pathways di LS (Fig.1B).
Progettazione e sintesi di inibitori di RAD52
Per sviluppare ulteriormente l'approccio terapeutico proposto, ci siamo concentrati sull'osservazione che nelle cellule proliferanti, quando il meccanismo convenzionale guidato da BRAC2/RAD51 è inibito, può avere luogo una via di HR alternativa, dipendente da RAD52. Questo approccio alternativo può proteggere le cellule tumorali difettose di BRCA1/2 dall'effetto sintetico letale dell'inibizione di PARP, indicando RAD52 come un interessante bersaglio terapeutico per le forme tumorali difettose di BRCA2. Per questi motivi, si può ipotizzare che l'inibizione simultanea di RAD52, PARP e dell'interazione RAD51/BRCA2 possa portare a una robusta LS a tre vie nelle cellule tumorali che mantengono la funzione BRCA. Pertanto, progetteremo e sintetizzeremo nuovi inibitori di RAD52 e ne caratterizzeremo le proprietà chimiche. Infine, i composti saranno testati in modelli cellulari di cancro al pancreas.
Membri del Laboratorio
Marinella Roberti, Professoressa Associata
Greta Bagnolini, Assegnista di ricerca (progettazione e sintesi di piccole molecole)
Progetti di internato (2)
Due posizioni di stage sono disponibili ogni anno per laureandi. Gli studenti contribuiranno alla progettazione razionale delle molecole, considerando la loro fattibilità sintetica e con il supporto di metodologie computazionali. Gli studenti effettueranno ricerche di letteratura utilizzando database online per tenersi aggiornati sul loro campo di ricerca. Inoltre, discuteranno i risultati scientifici con altri membri del gruppo che lavorano sullo stesso progetto.
Pubblicazioni significative
- Previtali V, Bagnolini G, Ciamarone A, Ferrandi G, Rinaldi F, Myers SH, Roberti M, Cavalli A. (2024). New Horizons of Synthetic Lethality in Cancer: Current Development and Future Perspectives. J Med Chem. Vol. 67, p. 11488-11521. doi: 10.1021/acs.jmedchem.4c00113.
- Myers SH, Poppi L, Rinaldi F, Veronesi M, Ciamarone A, Previtali V, Bagnolini G, Schipani F, Ortega Martínez JA, Girotto S, Di Stefano G, Farabegoli F, Walsh N, De Franco F, Roberti M, Cavalli A. (2024). An 19F NMR fragment-based approach for the discovery and development of BRCA2-RAD51 inhibitors to pursuit synthetic lethality in combination with PARP inhibition in pancreatic cancer. Eur J Med Chem. Vol. 265, Article number 116114, doi: 10.1016/j.ejmech.2023.116114
- Bagnolini,G., Balboni, B., Schipani, F., Gioia ,D., Veronesi , M., De Franco , F,. Kaya , C., Jumde , R.P., Ortega ,J.A., Girotto , S., Hirsch , A.K.H., Roberti , M., Cavalli, A. (2022). Identification of RAD51-BRCA2 Inhibitors Using N-Acylhydrazone-Based Dynamic Combinatorial Chemistry. ACS Med Chem Lett. Vol. 13, 1262-1269, doi: 10.1021/acsmedchemlett.2c00063
- Bagnolini G., Milano D., Manerba M., Schipani F., Ortega J. A., Gioia D., Falchi F., Balboni A., Farabegoli F., De Franco F., Robertson J., Pellicciari R., Pallavicini I., Peri S., Minucci S., Girotto S., Di Stefano G., Roberti M., Cavalli A. (2020). Synthetic Lethality in Pancreatic Cancer: Discovery of a New RAD51-BRCA2 Small Molecule Disruptor That Inhibits Homologous Recombination and Synergizes with Olaparib. JOURNAL OF MEDICINAL CHEMISTRY, vol. 63, p. 2588-2619, doi: 10.1021/acs.jmedchem.9b01526
- Roberti, M., Schipani, F., Bagnolini, G., Milano, D., Giacomini, E., Falchi, F., Balboni, A., Manerba, M., Farabegoli, F., De Franco, F., Robertson, J., Minucci, S., Pallavicini, I., Di Stefano, G., Girotto, S., Pellicciari, R., and Cavalli, A. (2019). Rad51/Brca2 Disruptors Inhibit Homologous Recombination and Synergize with Olaparib in Pancreatic Cancer Cells. EUROPEAN JOURNAL OF MEDICINAL CHEMISTRY, vol. 165, p. 80-92, doi: 10.1016/j.ejmech.2019.01.008
Contatti
-
Professoressa associata confermata
Dipartimento di Farmacia e Biotecnologie - FaBiT
Via Belmeloro 6
Bologna (BO)
Tel: +39 051 20 9 9738