Medicinal Chemistry - Chemical Biology. Coordinator: Roberti
Design and synthesis of novel biologically active small molecules, which can be used as putative lead candidates or tools for the discovery of new therapeutically relevant targets.
Research themes
The main research topics concern the design and synthesis of small molecules able to modulate molecular target involved in cancer development.The rational design of new molecules is carried out in collaboration with the computational pharmaceutical chemistry group (Prof. M. Masetti and Prof. A. Cavalli). The synthetic strategies applied include, in addition to "classical" methods, a combinatorial chemistry approach, working in parallel to obtain small libraries of drug-like molecules that are characterized by spectroscopic and spectrometric techniques. In addition, microwave-assisted synthesis is carried out according to a sustainable chemistry. In the following the two main projects are described.
Design and synthesis of disruptors of Rad51/BRCA2 interaction
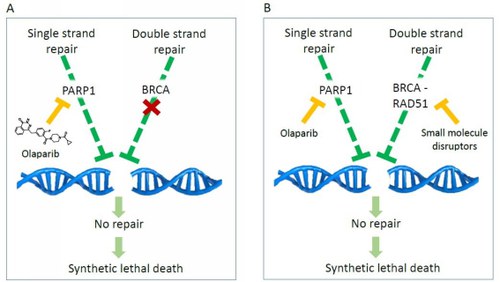
The Rad51/BRCA2 interaction is crucial for repairing damage to the double helix of DNA (homologous recombination (HR)). Since many antitumor drugs cause damage to the double strand of DNA, inhibiting HR can increase the sensitivity of cancer cells to these chemotherapeutics and other DNA damage repair inhibitors, such as PARP inhibitors.Olaparib was the first PARP inhibitor approved in 2014 for the treatment of breast cancer and pancreatic tumors with non-functioning BRCA2. In these patients, inhibiting PARP simultaneously compromises two independent mechanisms of DNA repair, thus inducing synthetic lethality (SL) and death of cancer cells. (Fig. 1A)The new inhibitors Rad51-BRCA2 can therefore be studied as chemosensitizing agents in association with chemotherapeutics that damage DNA or radiotherapy. In particular, they could chemically compromise the functioning of BRCA2 in patients where it is not defective and in association with olaparib trigger a synthetic lethality totally generated by small molecules, opening up new avenues for PARP-based inhibitors. The new paradigm could also be extended to other pairs of genes involved in different pathways of SL (Fig.1B).
Design and synthesis of RAD52 inhibitors
To further develop the proposed therapeutic approach, we focused on the observation that in proliferating cells an alternative, RAD52-dependent HR pathway can take place when the conventional BRAC2/RAD51 driven mechanism is inhibited. This alternative approach can protect BRCA1/2 defective tumor cells from the synthetic lethal effect of PARP inhibition, indicating RAD52 as an attractive therapeutic target for the BRCA2 defective tumor forms. For these reasons, it can be hypothesized that the simultaneous inhibition of RAD52, PARP and RAD51/BRCA2 interaction may results in a robust three-pathway SL in tumor cells maintaining BRCA function. We will therefore design and synthesize novel RAD52 inhibitors, and characterize their chemical properties. Finally, the compound will be tested in pancreatic cancer cell models.
Lab Members
Marinella Roberti, Associate Professor
Greta Bagnolini, Research Fellow
Internship projects (2)
Two internship positions are available each year for undergraduates. The students will contribute to the rational design of the molecules, considering their synthetic feasibility and with the support of computational methodologies. The students will carry out literature search using online databases to keep updated about their research field. Moreover, they will discuss is scientific results with other members of the group, working on the same project.
Main publications
- Previtali V, Bagnolini G, Ciamarone A, Ferrandi G, Rinaldi F, Myers SH, Roberti M, Cavalli A. (2024). New Horizons of Synthetic Lethality in Cancer: Current Development and Future Perspectives. J Med Chem. Vol. 67, p. 11488-11521. doi: 10.1021/acs.jmedchem.4c00113.
- Myers SH, Poppi L, Rinaldi F, Veronesi M, Ciamarone A, Previtali V, Bagnolini G, Schipani F, Ortega Martínez JA, Girotto S, Di Stefano G, Farabegoli F, Walsh N, De Franco F, Roberti M, Cavalli A. (2024). An 19F NMR fragment-based approach for the discovery and development of BRCA2-RAD51 inhibitors to pursuit synthetic lethality in combination with PARP inhibition in pancreatic cancer. Eur J Med Chem. Vol. 265, Article number 116114, doi: 10.1016/j.ejmech.2023.116114
- Bagnolini,G., Balboni, B., Schipani, F., Gioia ,D., Veronesi , M., De Franco , F,. Kaya , C., Jumde , R.P., Ortega ,J.A., Girotto , S., Hirsch , A.K.H., Roberti , M., Cavalli, A. (2022). Identification of RAD51-BRCA2 Inhibitors Using N-Acylhydrazone-Based Dynamic Combinatorial Chemistry. ACS Med Chem Lett. Vol. 13, 1262-1269, doi: 10.1021/acsmedchemlett.2c00063
- Bagnolini G., Milano D., Manerba M., Schipani F., Ortega J. A., Gioia D., Falchi F., Balboni A., Farabegoli F., De Franco F., Robertson J., Pellicciari R., Pallavicini I., Peri S., Minucci S., Girotto S., Di Stefano G., Roberti M., Cavalli A. (2020). Synthetic Lethality in Pancreatic Cancer: Discovery of a New RAD51-BRCA2 Small Molecule Disruptor That Inhibits Homologous Recombination and Synergizes with Olaparib. JOURNAL OF MEDICINAL CHEMISTRY, vol. 63, p. 2588-2619, doi: 10.1021/acs.jmedchem.9b01526
- Roberti, M., Schipani, F., Bagnolini, G., Milano, D., Giacomini, E., Falchi, F., Balboni, A., Manerba, M., Farabegoli, F., De Franco, F., Robertson, J., Minucci, S., Pallavicini, I., Di Stefano, G., Girotto, S., Pellicciari, R., and Cavalli, A. (2019). Rad51/Brca2 Disruptors Inhibit Homologous Recombination and Synergize with Olaparib in Pancreatic Cancer Cells. EUROPEAN JOURNAL OF MEDICINAL CHEMISTRY, vol. 165, p. 80-92, doi: 10.1016/j.ejmech.2019.01.008
Contacts
-
Associate Professor
Dipartimento di Farmacia e Biotecnologie - FaBiT
Via Belmeloro 6
Bologna (BO)
Tel: +39 051 20 9 9738